Interdisziplinäres Darmzentrum
Haben Sie als Darmkrebserkrankter oder Darmpolypenträger Angehörige, so besitzen diese ein erhöhtes Risiko, auch an Darmkrebs zu erkranken.
 Aus diesem Grund sollten Familien dieses erhöhte
Risiko frühzeitig realisieren und eine adäquate Darmkrebsvorsorge durchführen.
Aus diesem Grund sollten Familien dieses erhöhte
Risiko frühzeitig realisieren und eine adäquate Darmkrebsvorsorge durchführen.
Prinzipiell unterscheidet man zwischen erblichen Darmkrebserkrankungen sowie familiär gehäuften Erkrankungen. Bei etwa 25 % der Patienten mit Dickdarmkrebs findet man eine familiäre Häufung
der Erkrankung. Bei etwa 5 % der Patienten liegt eine der bekannten erblichen Formen von Dickdarmkrebs vor. Diese bekannten erblichen Tumorerkrankungen umfassen den erblichen Dickdarmkrebs ohne
Auftreten multipler Darmpolypen, welcher als „Hereditary Non- Polyposis Colorectal Cancer“ = HNPCC oder Lynch-Syndrom bezeichnet wird und die erblichen Polyposis (Auftreten multipler Darmpolypen)
-Syndrome. Hierzu gehören die Familiäre adenomatöse Polyposis (= FAP) sowie andere seltenere Syndrome.
Sind mehrere Personen aus einer Familie an einem kolorektalen Karzinom oder anderen Tumoren erkrankt, sollte der Verdacht auf eine erbliche Darmkrebserkrankung geäußert werden.
Auch die Entwicklung von mehrfachen Tumoren bei einem Patienten oder eine Einzelerkrankung bei einem jungen Patienten können auf erblichen Darmkrebs hinweisen. Ziel genetischer
Untersuchungen ist es, Risikopersonen zu identifizieren und einem engmaschigen Früherkennungsprogramm zuzuführen. Risikopersonen hingegen, bei denen die Veranlagung zur Tumorerkrankung
ausgeschlossen wurde, können aus dem Früherkennungsprogramm entlassen werden, da sie kein höheres Tumorrisiko als die Allgemeinbevölkerung haben.
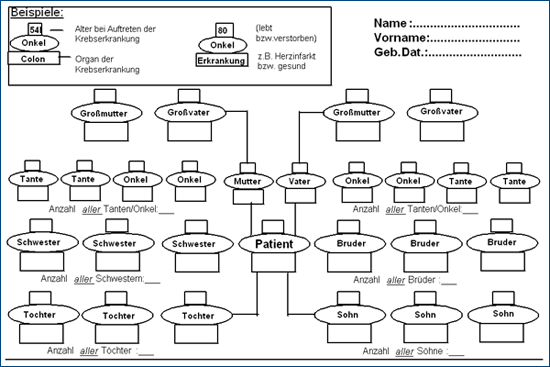
Für die einzelnen erblichen Darmkrebserkrankungen sind Veränderungen (Mutationen) in verschiedenen Genen bekannt. Bei Verdacht auf eine erbliche Tumorerkrankung in der Familie sollte eine
humangenetische Beratung empfohlen werden. Nach Anfertigung einer ausführlichen Anamnese inklusive Stammbaumanalyse und der Dokumentation der aufgetretenen Tumorerkrankungen in der Familie
können heutzutage gezielte molekulargenetische Untersuchungen durchgeführt werden.
Angeborener Nicht-Polypöser Dickdarmkrebs (HNPCC)
Die häufigste Form des erblichen Darmkrebses ist das HNPCC-Syndrom (Hereditary Non-Polyposis Colorectal Cancer = hereditäres kolorektales Karzinom ohne Polyposis), das durch das Auftreten von
insbesondere Dick- und Enddarmkrebserkrankungen, an zweiter Stelle Gebärmutterkrebserkrankungen und seltener Tumorerkrankungen am Magen, Dünndarm, Bauchspeicheldrüse, Eierstock, ableitende
Harnwege, Haut und Hirn gekennzeichnet ist. HNPCC wird autosomal- dominant vererbt, d.h. die Mutation wird auf 50% der Nachkommen übertragen. Für Mutationssträger besteht ein ca. 70-80%iges
Erkrankungsrisiko für Tumorerkrankungen des Dick- oder Enddarms und ein ca. 40%iges Risiko für Tumorerkrankungen der Gebärmutter. Die anderen oben genannten Tumorerkrankungen treten wesentlich
seltener auf. Grundsätzlich treten Tumorerkrankungen in früheren Lebensjahren auf, in der Regel aber nicht vor dem 20. Lebensjahr.
Molekulargenetischer Hintergrund der HNPCC-Diagnostik
Keimbahnmutationen in den DNA-Mismatch-Reparaturgenen MLH1, MSH2, MSH6 und PMS2 sind die molekulargenetische Ursache für ein HNPCC-Syndrom. Am häufigsten werden Mutationen im MLH1 und
MSH2-Gen nachgewiesen. Die Tumoren von HNPCC-Patienten zeigen charakteristischerweise (ca. 80-90%) eine hochgradige Mikrosatelliteninstabilität. Mikrosatelliten sind kurze repetitive
DNA-Sequenzen, die sehr anfällig sind für Fehler, die während der Replikation der DNA auftreten können. Normalerweise werden diese Fehler durch ein intaktes DNA Mismatch-Reparatursystem repariert.
Besteht ein Funktionsausfall eines dieser DNA-Mismatch-Reparaturgene im Tumor, können die Fehler nicht korrigiert werden und es kommt zum Auftreten zusätzlicher Allele, was als
Mikrosatelliteninstabilität bezeichnet wird. Mikrosatelliteninstabilität kann jedoch auch in sporadisch auftretenden Kolonkarzinomen vorkommen (ca. 15%). Bei der Mikrosatelliten-analyse
werden 5 Standardmikrosatellitenmarker untersucht. Zeigen mindestens 2 Marker eine Instabilität liegt eine hochgradige Mikrosatelliteninstabilität vor und es wird die Durchführung einer
Keimbahnmutationsanalyse empfohlen. Zeigt nur ein Marker eine Instabilität wird eine erweiterte Mikrosatellitenuntersuchung mit einer Analyse von fünf weiteren Markern durchgeführt.
Die Untersuchung auf Mikrosatelliteninstabilität bei Verdacht auf HNPCC (Erfüllung der Bethesda-Kriterien) ist eine Präscreening-Untersuchung, die die Durchführung der anschließenden
Mutationsanalysen auf die Patienten mit einem hochgradig instabilen Tumor beschränkt.
Parallel zur Mikrosatellitenanalyse wird eine immunhistochemische Untersuchung der Expression der 4 DNA-Reparaturgene im Tumor des Patienten durchgeführt. Das DNA-Reparaturgen, das bei
HNPCC Patienten eine Keimbahnmutation aufweist, zeigt charakteristischerweise im Tumor des Patienten einen Expressionsverlust. Die immunhistochemische Untersuchung kann somit auf das Gen
hinweisen, in dem die Keimbahnmutation präferentiell liegen könnte und erleichtert somit die weitere molekulargenetische Diagnostik. Nur durch den Nachweis einer Keimbahnmutation in einem
der DNA-Mismatchreparaturgene wird die Diagnose eines HNPCC-Syndromes molekulargenetisch gesichert und es ergibt sich dann die Möglichkeit der prädiktiven Diagnostik von Familienmitgliedern,
die noch nicht erkrankt sind.
Familienanamnestische Eingangskriterien zur molekularen Abklärung auf das Vorliegen eines HNPCC-Syndromes
Amsterdam-II-Kriterien
1 HNPCC-assozierte Tumoren: Endometrium, Magen, Ovar, Pankreas, Dünndarm, Ureter und Nierenbecken, Gallengang, Gehirn (üblicherweise Glioblastome wie beim Turcot-Syndrom), Talgdrüsenadenome und Keratoakanthome (beim Muir-Torre-Syndrom). 2 Vorliegen von Tumor-infiltrierenden Lymphozyten, Crohn-ähnlicher lymphozytärer Reaktion, muzinöser/Siegelring-Differenzierung, oder medullärem Wachstum
Je nach Ergebnis des molekulargenetischen Tests sollte Patienten und Familien mit einem HNPCC-Syndrom ein entsprechendes Tumorfrüherkennungsprogramm, das im Rahmen einer humangenetisch- klinischen Beratung erläutert wird, angeboten werden. Folgendes Früherkennungsprogramm wird für Personen mit erhöhtem Risiko empfohlen:
Alter Untersuchung Häufigkeit ab dem 25. Lebensjahr (bzw. 5 Jahre vor dem frühesten Erkrankungsalter in der Familie) Körperliche Untersuchung einmal jährlich Ultraschalluntersuchung des Bauches einmal jährlich Komplette Darmspiegelung einmal jährlich Gynäkologische Untersuchung auf Gebärmutter- und Eierstockkrebs einmal jährlich ab dem 35. Lebensjahr Magenspiegelung einmal jährlich + obenFamiliäre adenomatöse Polyposis (FAP) Die zweithäufigste Form des erblichen Darmkrebses ist die „Familiäre adenomatöse Polyposis“ (FAP). Sie ist durch das Auftreten von Hunderten von Polypen im gesamten Dickdarm gekennzeichnet. Das Polypenwachstum beginnt meist im 2. Lebensjahrzehnt am Übergang zwischen Dick- und Enddarm. Im Alter von ca. 40 Jahren entwickelt sich mit fast 100%iger Wahrscheinlichkeit ein Darmkrebs, wenn die Polypen nicht rechtzeitig entfernt wurden. Einige Patienten entwickeln zusätzlich auch Tumore außerhalb des Dickdarms (z.B. im Duodenum, im Magen, in Knochen). Bei etwa 85% der Patienten beobachtet man zudem eine charakteristische Veränderung der Netzhaut. FAP wird autosomal- dominant vererbt und durch eine Mutation im Tumorsuppressorgen APC verursacht, die bei 80% der Patienten mit typischer FAP nachgewiesen werden kann. Beim FAP- Syndrom wird für Personen mit erhöhtem Risiko folgendes Früherkennungsprogramm empfohlen: Alter Untersuchung Häufigkeit ab dem 10. Lebensjahr Körperliche Untersuchung einmal jährlich Enddarm-Spiegelung (bei Beobachtung erster Polypen komplette Darmspiegelung) einmal jährlich Ultraschalluntersuchung des Bauches einmal jährlich Augenärztliche Untersuchung auf Veränderungen der Netzhaut einmalig vor der Entfernung des Dickdarms bzw. spätestens ab dem 30. Lebensjahr Erste Magen- und Zwölffingerdarm-spiegelung, danach - bei Nachweis von Adenomen - ohne Nachweis von Adenomen einmal jährlich alle drei Jahre


+49 (89) 4140-1260
Mo und Di: 16-18 Uhr,
Mi - Fr: 10-12 Uhr
Unsere Experten helfen Ihnen gerne weiter
.



„Eine kleine Drüse mit großer Wirkung“ – am 16. November 2023 ist Weltpankreaskrebstag

Bayerns Staatsminister für Gesundheit und Pflege Klaus Holetschek bedankt sich für Top Medizin






Klinikum rechts der Isar kooperiert mit Klinik in Ghana



 erhält Young Investigator Grant 2023 der IAP und der APA Foundation für Forschung
erhält Young Investigator Grant 2023 der IAP und der APA Foundation für Forschung 





